



諮詢:吳庶忠教授(香港科技大學生命科學部客座教授)[1]
整理:文麗兒(明光社項目主任)[2]
美國某幾間大學在新學年容許跨性別新生在入學註冊時自行選擇性別,西方國家不少高校亦因跨性別學生要求使用異性的洗手間,而須對簿公堂。在今天這個光怪陸離的世界,面對性別議題所引致的影響似乎是不能避免的趨勢,但除了政治正確及潮流大勢,處理性別議題更須要回到根本──從生物學認識性別。
性別之本──基因
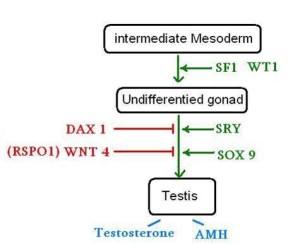
人類的性別決定系統(sex determination system)基本上可透過第23對染色體(即XX及XY)識別男與女。在男性獨有的Y染色體中有名為TDF(testis-determining factor),又名SRY[sex-determining region Y (SRY) protein]的基因連結蛋白(參圖一)。簡單來說,SRY是其中一個令胚胎在成長過程中發展成男性的主要的基因/蛋白,SRY的出現會命令細胞製造睪酮 (testosterone) 及抗穆氏管荷爾蒙(Anti-Müllerian hormone, AMH),以發展出男性生殖器官(參圖二)。

圖一
圖二
雖然SRY是其中一個主要控制胚胎順利發展成男性的基因蛋白,但它必須與其他相關而重要的基因/蛋白複合,才能令發展成為男性的過程順利完成。而不同的基因/蛋白其實互為影響,互相緊扣,因而其中一部分出現變異或錯誤編碼,都會引致接續的步驟出現錯誤。
SF1(steroidogenic factor 1)負責控制胚胎及青春期的性發展,當SRY與SF1複合就會上調成為轉錄因子SOX-9[SRY (sex determining region Y)-box 9],SOX-9在發展成男性的過程中都有著舉足輕重的作用,通過與SF1合作,SOX-9會製造塞特利氏細胞(Sertoli cells)中的AMH,以抑制女性生殖系統的發展。同時SOX-9會激活FGF9(Fibroblast growth factor 9)及PGD2(Prostaglandin D2),分別與它們形成一個前遺迴路(feedforward loop)的狀態,即令對方都能維持穩定的數量。
PGD2的功能是協助SOX-9保持足夠高的數量以激活其他基因;FGF9的功能是確保男性的健康發育,包括製造睪丸素及增加Sertoli cells;而Sertoli cells的作用是分泌不同的物質,包括:AMH於胚胎期釋出用以抑制女性生殖系統發展;ABP(androgen binding protein)用以增加在睪丸內的精細管的睪酮濃度以刺激精子製造;雌二醇(estradiol,17β-estradiol)是由Sertoli cells內的芳香酶(aromatase)直接把睪酮轉化成為17β-estradiol而製造精子等等。
假如SOX-9或相關的基因出現變異(如 SOX-9 deletion),便會引起性別逆轉甚至雌雄同體(46XY female due to SOX-9 deletion, 名Campomelic dysplasia);假如FGF9及Dax-1缺失,即使有XY染色體的胚胎都會變成女性。同樣如SRY出現不正常的活動情況都會引致雌雄同體的情況。
從雙性別的胚胎到單一性別的嬰兒
人類不能否認男女性別的發展是由基因主宰,基因是一個極之複雜而且充滿奧秘的領域,我們嘗試從暫時已知道的實況去理解基因的運作及其影響。完全發展的生殖系統除了基因性別[Chromosomal (genetic) sex],還包括了性腺性別(Gonadal sex,即內生殖器,包括男性的睪丸、輸精管;女性的卵巢、子宮、輸卵管等)及表型性別(Phenotypic sex,即外生殖器,包括男性的陰莖、女性的陰核等)。
大部份人(99.99%)都有一套特定的染色體(23組共46條),而每一條染色體都由2組DNA合成,一組來自父親,一組來自母親,所以每一組染色體都帶著父母二人的基因。開首的22組染色體在男與女都沒有太大分別,但第23組染色體,即一般所理解的性染色體,則有很大的不同。男的性染色體為XY(X來自母親,Y來自父親),女的則為XX(X來自母親,X來自父親)。一般來說,受精卵發展至胚胎的初期,體內都有兩組潛在的生殖系統(Wolffian system及Müllerian system),所以暫時未能開始分辨為男性或女性。
直至第6至8周,如胚胎第23組染色體為XY (即男性),在Y染色體內的SRY開始與其他的基因蛋白進行複合,並發展出不同的狀態(如上文對於基因的描述),釋出AMH以抑制女性生殖系統的發育;並令男性的生殖系統持續發育至成熟,因此在嬰兒出生時可以看到已發展的男性外生殖器(參圖三)。一旦基因表達出現嚴重錯失,如SOX9 deletion,便會引伸出雌雄同體的情況。

圖三
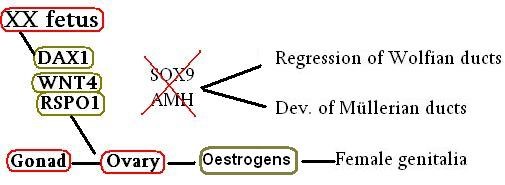
如胚胎第23組染色體為XX(即女性),在X染色體中的Dax1的角色十分重要,它的其中一個功能是抗衡SRY以抑制它與其他基因蛋白進行複合,防止男性的生殖系統及製造AMH (參圖四);另外,亦是控制製造荷爾蒙的組織(包括:腎上腺、腦下垂體、下丘腦及睪丸與卵巢)的發育。當Dax1/NR0B1基因出現變異便會引致先天性腎上腺發育不良(X-linked congenital adrenal hypoplasia, X-linked AHC) 及性腺機能減退 (hypogonadotropic hypogonadism,HH)。[3] X-linked AHC最主要的影響是腺體發育不完全,對男性的主要影響是導致缺乏男性荷爾蒙,令生殖器官發育不完全、隱睪症(cryptorchidism)的情況出現、青春期較遲出現、不育等。女性相對較少受到影響,但亦有女性因患上X-linked AHC而缺乏女性荷爾蒙、青春期較遲出現及沒有月經。而HH患者的腦下垂體無法釋放促性腺激素釋放素 (Gonadotropin-releasing hormone, GnRH) ,缺乏GnRH則無法分泌促性腺激素 (gonadotropins)包括促卵泡激素(follicle-stimulating hormone, FSH)及促黃體素(luteinizing hormone, LH)。
圖四
簡單來說,FSH與LH的功能是調節身體的發育、生長、青春期成熟和生殖過程,刺激卵巢內未成熟的卵子的成長及刺激精母細胞分裂以製造精子。當FSH及LH無法正常分泌時會影響睪酮及雌激素的製造。過少的FSH會引致性腺功能減退(hypogonadism),最明顯的症狀是男性未能製造正常數目的精子,女性的生理周期終止等。
基因變異的不幸──雙性人
誠然,生命的成長確實是一個奧秘,由天生擁有男女兩性的生殖系統的胚胎一直發展成為單一性別胎兒,如相關的基因出現變異,一般會引致性發展障礙(disorders of sex development, DSD),嚴重的情況會引致雙性人(intersex),為整個生命帶來逆轉。
值得注意的是DSD與其他在DSM-5中的精神疾病所提及的性功能障礙(sexual dysfunctions) 有所不同。性功能障礙一般是指因心理因素或環境因素而引致性功能失調或障礙,但DSD所指的障礙乃因基因變異而引致的疾病 (genetic disorder)。因此,如要以性別光譜論解釋生理層面的兩性分野並不合適。生理性別不是光譜,沒有程度之分,一般基因得到正常發展的人不會說自己的生理構造有七成是男性,三成是女性,而是百份百是男或女。至於因基因變異而生理性徵沒有得到正常發展,即一些患上DSD的人士,身體結構會同時擁有兩個性別的性徵,無法單一地劃分為男性或女性。在這樣的情況下,社會需要考慮的是如何支援面對DSD的人士,令他們能夠得到適當的保障及治療,而不應與其他政治運動的訴求混淆。
注釋
[1] 吳庶忠教授於美國取得生物化學哲學博士學位,在著名科學期刊出版逾百篇「同行評審」論文,包括Science和Nature。吳教授現時是香港科技大學生命科學部成員,教授本科生及學士後生物科技課程。
[2] 鳴謝明光社供稿。原刊於明光社網站:http://www.truth-light.org.hk/article/title/n5616,2015年11月5日。
[3] hypogonadotropic hypogonadism,HH;https://www.nlm.nih.gov/medlineplus/ency/article/000390.htm.